In LCAO, it is the set of atomic orbitals (AOs) that is the basis, and the coefficients are the basis expansion coefficients. For example, take the hydrogen molecule, with 1 atomic orbital on each atom:
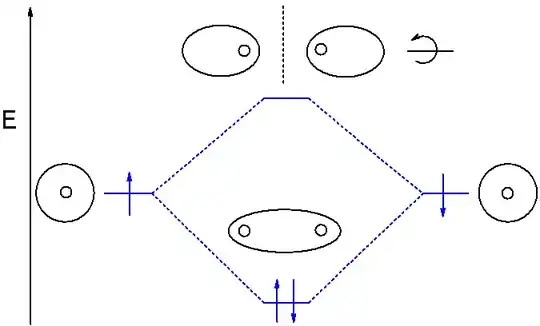
The lower energy MO, the bonding one, will be (excluding normalization):
$$
\psi_{\mathrm{1s}} = \phi_{\text{left}} + \phi_{\text{right}}
$$
and the higher energy MO, the antibonding one, will be (excluding normalization):
$$
\psi_{\mathrm{1s}^{*}} = \phi_{\text{left}} - \phi_{\text{right}}
$$
This means that for the bonding MO, $c_{\text{left}} = 1$ and $c_{\text{right}} = 1$, while for the antibonding MO, $c_{\text{left}} = 1$ and $c_{\text{right}} = -1$. If your basis was different (say, the atomic orbitals were of different radial extents for the two atoms), then the coefficients would be different to accommodate that. They aren't part of the basis set.
Since you tagged this with computational-chemistry, I assume you also want to know about basis sets in computational chemistry. Conceptually, they are identical to LCAO-MO bases, but what may be confusing is that each atomic orbital itself may be composed of multiple functions (called primitive functions), rather than just the single function for each AO as seen above. This leads to another set of coefficients, called contraction coefficients, describing how the primitive functions are linearly combined to form a contracted function.
An "implementation" detail is that the functional form of AOs is usually Gaussian functions, which have a parameter in the exponent (here, $\alpha$):
$$
\phi_{r}(x) = e^{-\alpha x^2}
$$
This means that to fully define an atomic orbital basis composed of contracted Gaussian-type orbitals (CGTOs), one needs both the contraction coefficients and exponents. They usually look like the following. For each angular momentum type, there may be one or more contracted functions, each composed of one or more primitives. For example, in the hydrogen definition below, there are 2 functions to describe s orbitals, one of which has three primitives and the other is not contracted. The contraction coefficients are in the first column and the exponents in the second column.
****
H 0
S 3 1.00
13.0107010 0.19682158E-01
1.9622572 0.13796524
0.44453796 0.47831935
S 1 1.00
0.12194962 1.0000000
****
C 0
S 5 1.00
1238.4016938 0.54568832082E-02
186.29004992 0.40638409211E-01
42.251176346 0.18025593888
11.676557932 0.46315121755
3.5930506482 0.44087173314
S 1 1.00
0.40245147363 1.0000000
S 1 1.00
0.13090182668 1.0000000
P 3 1.00
9.4680970621 0.38387871728E-01
2.0103545142 0.21117025112
0.54771004707 0.51328172114
P 1 1.00
0.15268613795 1.0000000
D 1 1.00
0.8000000 1.0000000
****
The last note is that when performing a Hartree-Fock or similar calculation, the contraction coefficients and the exponents don't change. That would mean the basis set changes. The basis set is fixed, it is the MO coefficients that change, the same coefficients that appear in the LCAO-MO equations.
Image taken from here. Basis set is Def2-SV(P).
{kind=link}