The Next-Gen sequencers cannot sequence a very long stretch of DNA with good reliability (~150 for the recent model- HiSeq2000; even less for older models such as GA (40), GA-II (70), GA-IIx (90)). For increasing the confidence in a certain hit, it was sequenced from both the ends.
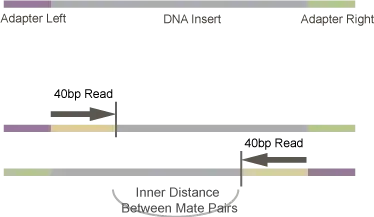
For example, if you have selected 500bp DNA fragment, then after ligating adapters to both the ends, it is sequenced from both the direction up to 150bp. This would leave an unsequenced "insert" region of 200bp. (In the example image below, they have sequenced up to 40bp [case of old GA] )
During assembly you stitch together the fragments of DNA to find out the larger DNA from where the fragments arise. In case of RNAseq, these arise from a transcript, and your assembly should give you the complete transcript (mRNA or ncRNA etc). There are two basic types of assembly: reference guided assembly and de-novo assembly. In the former you use a sequence such as the genome as a reference to assemble the transcripts. If such a reference is not available then you have to go for de-novo assembly.
The assembly algorithms use several parameters and since these are computer algorithms and not some kind of magic, their output depends to an extent on the different parameters.
In case of paired end data there are some parameters that are important. Most important is the size of the insert. In case of a 500bp fragment, you'll end up with an unsequenced region of 200bp. This is not much of a problem with reference guided assembly because you can figure out the sequence of the insert based on where the sequenced region align to the reference. The average insert length is important to remove discordant reads (aligning too far apart in the reference). In case of de-novo assembly, the insert will remain unsequenced even if you know that the final transcript looks something like:
frag1-frag6-frag3-frag9-frag4
So, to get the sequence of the assembly, you need to sequence the insert regions. This is not a problem if you at least know the order of fragments in the assembly. However you should know the insert size to get the assembly size correct and as skyminge said, in scaffolding. Obtaining this insert length is not that difficult (You need not provide it as a parameter. Most algorithms can calculate it automatically).
Another parameter in de-novo assembly is k-mer length (the sequence reads are broken down into k-mers for better assembly). I cannot explain the algorithm of assembly here in detail. You can check the manuals/papers of common assembly algorithms like Velvet, SOAPdenovo, Euler [de novo]; cufflinks [reference based]
I have mentioned transcriptome sequencing here but the principles are same for genome sequencing too.
Back to your main question:
Why is assembling paired end illumina without any input parameters an important problem?
Because it is less effort; but tweaking may be difficult. I won't consider it as an important problem. There are other important algorithmic optimizations that are required with de-novo assembly.